Research Interests
We are a theoretical/computational research group that applies quantum mechanics to study chemical phenomena at the level of individual atoms. Our work involves both developing novel simulation methods and applying them to uncover general design principles that can enable the development of better catalysts, next-generation lighting devices, and novel materials. We frequently collaborate with experimental groups, combining our complementary toolkits to solve complex chemical problems and advance our shared scientific goals.
Our research is currently focused on the following areas:
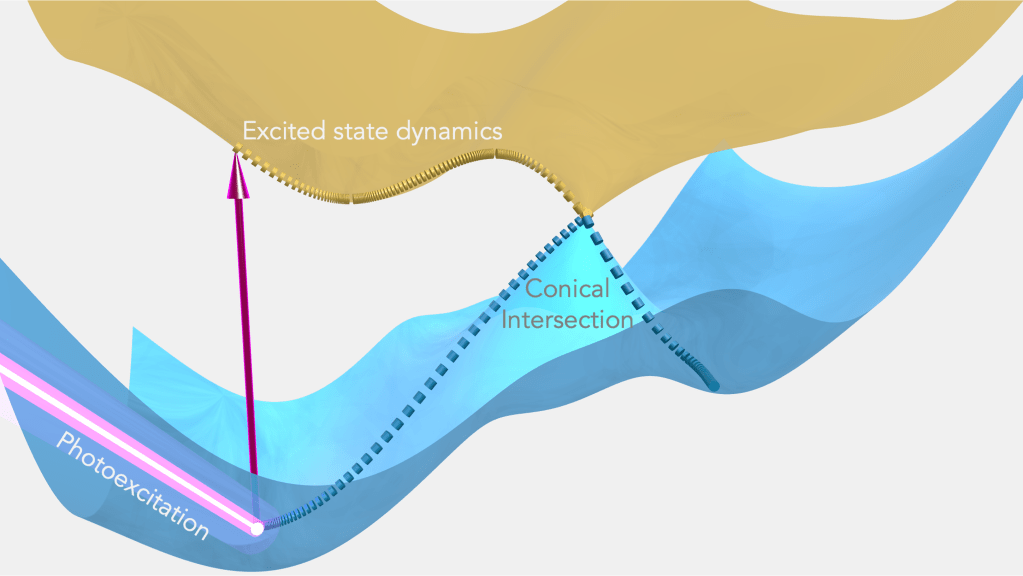
Simulating Electronic Excited States
Electronic excited states play a key role in the photophysics and photochemistry of chemical systems. The non-aufbau electronic configurations of electronic excited states make their simulation considerably more challenging than that of the ground state, which is a minimum of energy vs electronic degrees of freedom. This is especially the case for structures far away from the ground state equilibrium geometry (the “Franck-Condon region”), which are readily accessed by photoprocesses. For instance, population transfer between electronic states occurs most readily at geometries where the states are near degenerate in energy (“conical intersections”), leading to a breakdown of the Born-Oppenheimer approximation that decouples electronic and nuclear degrees of freedom. Accurate description of potential energy surfaces around these conical intersections is quite challenging for traditional electronic structure methods, with many popular approaches failing to even yield a qualitatively satisfactory description. We are working to develop new electronic structure methods that can accurately and efficiently simulate excited states, enabling the prediction of photophysical, photochemical, and spectroscopic properties.
Computational Photocatalyst Design
Light absorption excites chemical systems into nonequilibrium atomic configurations on excited state potential energy surfaces. This can induce chemical reactivities, such as ultrafast bond cleavage or charge separation, that are difficult to access via thermal processes in the ground state. Photocatalysis is therefore an attractive route for efficient synthesis of products that would otherwise require extreme conditions in the ground state. However, effective bimolecular photocatalysis can only occur if the catalyst excited state lives long enough to encounter a substrate. It is therefore critical to uncover and block potential pathways for a photocatalyst excited state from relaxing to the ground state. We are working to computationally uncover the design principles for effective photocatalysis with both main group species and transition metal complexes, in order to design more selective and energy efficient catalysts.

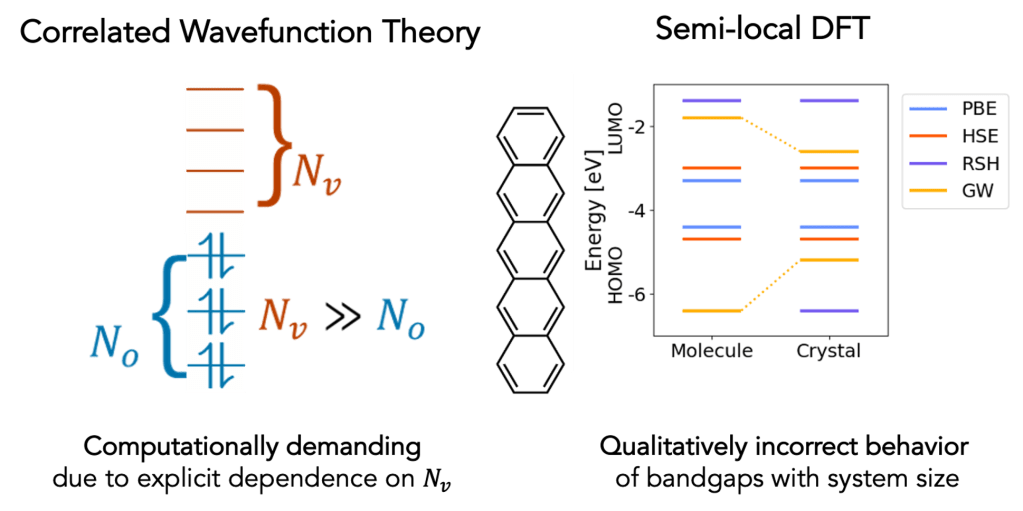
Efficiently Modeling Nonlocal Correlation
Density functional theory (DFT) is widely utilized to model the electronic structure of chemical systems, as it is computationally efficient compared to methods directly simulating the many-electron wavefunction. Most existing density functionals describe electron correlation using a local approach, i.e. utilizing information about the electron density at a single point in space independent of all other points. As a result, DFT is typically challenged in describing long-range correlations between electrons and related many-body effects, such as condensed phase screening of bandgaps. Correlated wavefunction methods like coupled cluster theory can describe nonlocal electron correlation, but only at prohibitive computational cost due to explicit dependence on unoccupied molecular orbitals. This makes use of such methods challenging for large condensed phase problems, precisely where the effects of long-range electron correlation matter the most. Our group develops efficient models for nonlocal correlation that seamlessly integrate with density functional theory, enabling more accurate simulations of condensed-phase systems.
